AlphaFold: Protein-Structure Prediction with AI
Written By: Braeden Cullen
Protein structure prediction is an important aspect of computational biology and is the process by which three-dimensional structures of proteins are predicted based on their amino acid sequence. Accurate protein structure prediction is of unparallel importance due to its wide-ranging implications in the field of biology. This process enables the successful development of drugs by helping researchers determine which molecules can effectively bind to the protein and predict the nature of these interactions (Deng, H., et al.). Prior to the developments made during this past year, Biologists determined protein structures through experimental procedures, which are costly and time-consuming. Computational techniques, although becoming increasingly common, were not viable due to many having critical weaknesses or lackluster accuracy. The newest machine learning model, AlphaFold, developed by a team of researchers from DeepMind has effectively challenged this axiom. AlphaFold effectively leverages a complex algorithm to formulate highly accurate protein structure predictions within a reasonable timeframe. Through the distribution of AlphaFold to researchers worldwide, DeepMind hopes to revolutionize the field of Biology and make protein structure prediction increasingly accessible.
Past Developments
Computational biologists have been attempting to develop increasingly efficient and accurate methods for predicting three-dimensional protein structures since the late 20th century. Notable advances in this technology include the development of the Chou-Fasman method, which relied heavily on the probability parameters of the relative frequency of each amino acid’s appearance in each secondary structure (Chou-Fasman Algorithm). This method achieved an accuracy of roughly 60% (Chen, H., et. al). Another notable leap in accuracy came with the first implementation of machine-learning models to predict protein structures. Researchers utilized a neural network to identify common arrangements of secondary structures that were associated with specific proteins. This model was able to accurately predict approximately 70% of protein structures but had certain limitations that limited its effectiveness (Torrisi, M., et al.). Although both of these developments were quite impressive, even monumental, at the time, their accuracies pale in comparison to modern methods.
What Makes AlphaFold Unique
DeepMind has yet to release a paper documenting the exact technical aspects of AlphaFold, but at this point, the general consensus is that AlphaFold utilizes an attention network. An attention network is an exciting new development in the field of Artificial Intelligence that mimics the cognitive process of attention found in organisms. In short, this essentially means that the model works to identify discrete aspects of a larger problem, which are then pieced together to come up with a solution (Singh). The use of this updated model allows DeepMind to circumvent many of the issues that plagued previous AI-based protein folding models employed by opposing teams. The machine learning methods used at CASP13 and CASP14 vary greatly, according to the DeepMind team. This change resulted from an inability to increase the accuracy of the original AlphaFold model above roughly 70%, which led to the team completely altering how they approached the protein folding problem. This drastic change ultimately led to the creation of a model that achieved stunning accuracy, as can be seen in Figure 1. AlphaFold’s unique approach to the protein folding problem gave them an edge over other competitors’ models who frequently suffered from overfitting (Fang). Overfitting is the process by which a team’s model performs admirably on a training dataset but is unable to identify underlying commonalities between that data which causes it to perform poorly on new data. AlphaFold’s unique approach to the protein structure prediction problem paid off during the CASP14 competition, and it is highly likely that other competitors will also see a jump in performance as many begin to replicate the unique methods utilized by DeepMind into their own models. Furthermore, DeepMind also has access to incredibly high-performance computing systems which gave them an edge over other competitors who may have not been able to train their machine learning models as much or as efficiently as DeepMind due to computational limitations. AlphaFold was able to achieve a staggering increase in accuracy largely due to the employment of a new machine learning method and the immense computational resources they were allotted.
Figure 1
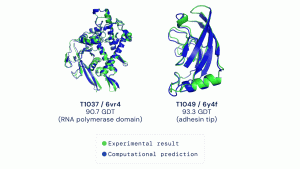
Image of Two Examples of AlphaFold’s Accuracy at Predicting Protein Structures
Source: DeepMind
How AlphaFold Is Already Revolutionizing the Field of Biology
The development of effective drugs relies heavily on accurate protein structure prediction. If researchers can determine the structure of a protein, they can simulate binding dynamics which is crucial for the development of effective drugs. Currently, drug development is severely bottlenecked by experimental protein structure prediction methods, which are extremely tedious and incredibly expensive. According to Steve Darnell, “The cost of solving a new, unique structure is on the order of $100,000.” (Darnell) In contrast, machine learning models can be run on relatively inexpensive computing equipment in a much shorter timeframe which amounts to a total cost that is orders of magnitude smaller than current methods. Currently, the DeepMind team has been utilizing AlphaFold to release structure predictions of understudied proteins associated with the SARS-CoV-2 virus (DeepMind). The publication of these structures is already enhancing researchers’ knowledge of the virus and is playing a role in the rapid development of treatments for the virus.
Conclusion
The incredible advancements made by DeepMind have the potential to revolutionize the field of biological research. According to Nobel prize-winning structural biologist Venki Ramakrishnan, the advancements made by DeepMind have “occurred decades before many people in the field would have predicted. It will be exciting to see the many ways in which it will fundamentally change biological research” (DeepMind). The immense benefits that come along with the development of DeepMind’s accurate model are already being realized as it is being applied to pressing issues such as the SARS-CoV-2 virus. In this situation, DeepMind was effectively utilized to predict proteins, notably the ORF8 protein, whose structures were previously undetermined. Out of nearly 180 million protein sequences currently known, only 170,000 protein structures are known due to the limitations of experimental methods of determining protein structure used prior to the development of AlphaFold (DeepMind). As AlphaFold continues to grow in popularity, we will continue to learn more about these unidentified proteins that could potentially have exciting functions that could prove useful in the development of new forms of medicine, alternative ways to manage the environment, and so much more.
Works Cited
Deng, H., Jia, Y., & Zhang, Y. (2018, July 20). Protein Structure Prediction. Retrieved December 26, 2020, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6407873
Singh, A. (2020, May 23). Attention Networks. Retrieved December 26, 2020, from https://towardsdatascience.com/attention-networks-c735befb5e9f
AlQuaraishi, M. (2019, May 22). AlphaFold at CASP13. Retrieved from https://ccsp.hms.harvard.edu/wp-content/uploads/2020/11/AlphaFold-at-CASP13-AlQuraishi.pdf
AlphaFold. (n.d.). Retrieved December 26, 2020, from https://deepmind.com/research/case-studies/alphafold
Chou-Fasman Algorithm. (n.d.). Retrieved December 26, 2020, from http://crdd.osdd.net/raghava/betatpred/chou.html
Chen, H., Gu, F., & Huang, Z. (2006, December 12). Improved Chou-Fasman method for protein secondary structure prediction. Retrieved December 26, 2020, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1780123/
Torrisi, M., Pollastri, G., & Le, Q. (2020, January 22). Deep learning methods in protein structure prediction. Retrieved December 26, 2020, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7305407/
Fang, J. (2019, July 05). Critical review of five machine learning-based algorithms for predicting protein stability changes upon mutation. Retrieved January 01, 2021, from https://academic.oup.com/bib/article/21/4/1285/5527140
Darnell, S. (2020, September 09). Why Structure Prediction Matters. Retrieved January 01, 2021, from https://www.dnastar.com/blog/structural-biology/why-structure-prediction-matters/
Computational predictions of protein structures associated with COVID-19. (n.d.). Retrieved January 01, 2021, from https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19